OpenFold3 Training Data Pipeline¶
Overview¶
This guide walks through preparing PDB structure data for OpenFold3 training. The pipeline converts raw PDB files into the preprocessed format consumed by the training script.
All output files directly required as an input to the training script are marked in bold with an asterisk (*). There is full flexibility in modifying the pipeline as long as it generates these required outputs, though it’s recommended to stick to the format below.
Tip
For detailed format specifications on the metadata and training/validation dataset caches, see the extended doc at Understanding Dataset Caches.
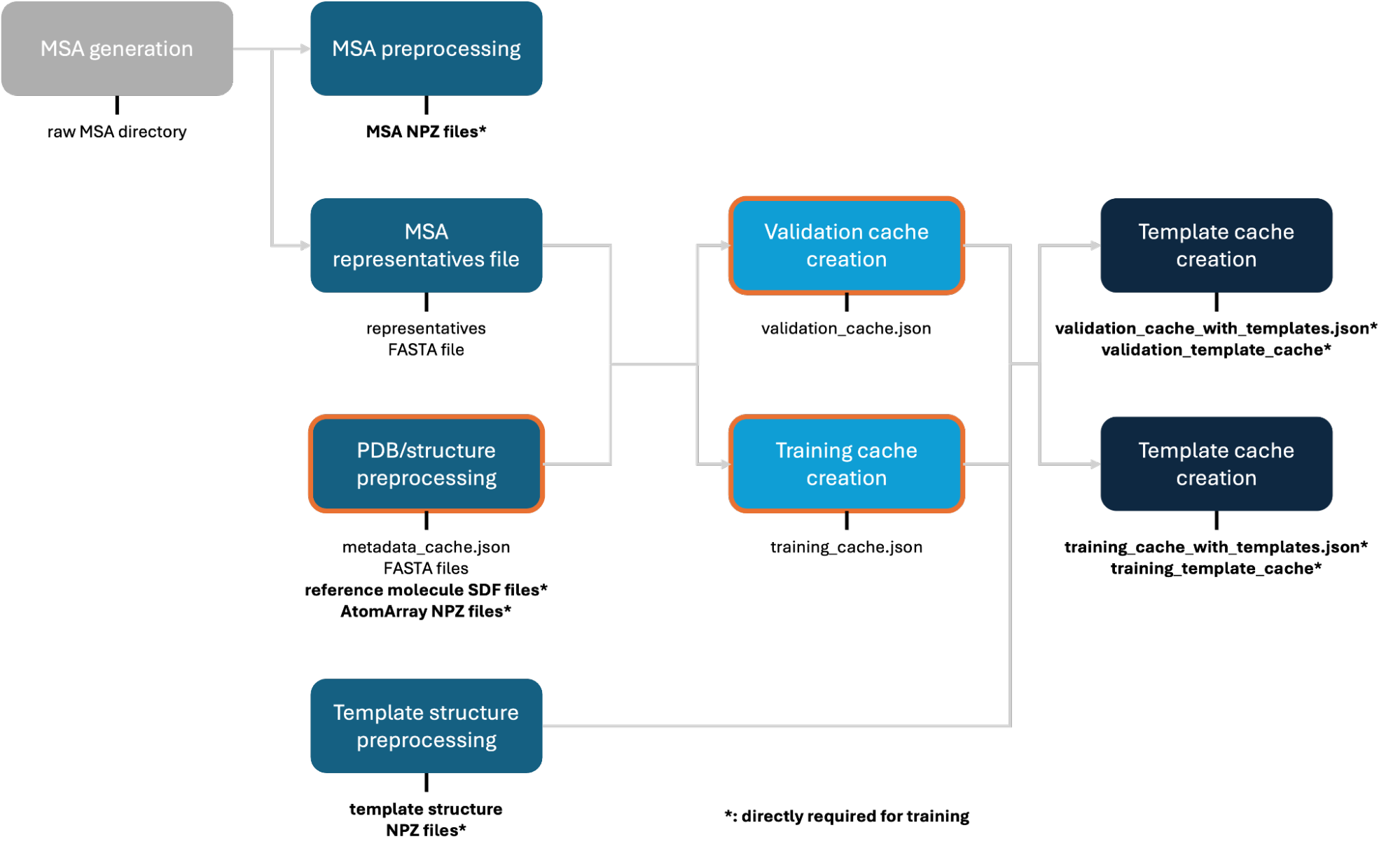
1. PDB Preprocessing¶
1.1 Structure Download¶
Our structure preprocessing expects a flat directory of .cif files. We provide an example script to generate this at scripts/download_pdb_mmcif.sh.
1.2 CCD Preprocessing (recommended but optional)¶
We employ Biotite for preprocessing, which relies on the Chemical Component Dictionary (CCD) for bond handling and structure parsing. By default it uses its own internal CCD copy, but when working with a specific PDB snapshot it can be cleaner to use the matching CCD version. This script prepares a CCD file for use with Biotite.
Script: scripts/data_preprocessing/preprocess_ccd_biotite.py
# Example usage with a fresh CCD
wget https://files.wwpdb.org/pub/pdb/data/monomers/components.cif.gz
gunzip components.cif.gz
python scripts/data_preprocessing/preprocess_ccd_biotite.py components.cif components.bcif
1.3 Structure Preprocessing¶
The core structure preprocessing converts raw PDB mmCIF files into an efficient .npz format storing Biotite AtomArrays, as well as a JSON index of all PDB contents. It performs the following steps:
Parsing structures using Biotite (bioassembly expansion, bond reading, chain renumbering to numerical IDs, entity/chain/molecule type assignment)
Cleaning up structures following the AlphaFold3 SI §2.5.4 protocol (residue conversions, removal of waters/hydrogens/clashing chains, adding unresolved atoms, etc.)
Generating reference molecules with RDKit conformers for each unique ligand (saved as SDF files*)
Extracting metadata into a
metadata.jsonwith structure-level, chain-level, and interface-level information
Script: scripts/data_preprocessing/preprocess_pdb_of3.py
Output: per-structure directories with NPZ*, FASTA, and optionally CIF files, plus a metadata.json* and reference molecule SDF files*:
structure_files/
├── 101m
│ └── 101m.cif
│ └── 101m.fasta
│ └── 101m.npz
├── 102l
│ └── 102l.cif
│ └── 102l.fasta
│ └── 102l.npz
├── ...
reference_molecules/
├── ATP.sdf
├── TRP.sdf
├── ...
metadata.json
2. Alignments¶
2.1 MSA Generation¶
AlphaFold3 training requires MSA generation against several sequence databases. This is computationally demanding and should be expected to take long for larger datasets.
Script: scripts/snakemake_msa/MSA_Snakefile
Instructions on how to run are in the MSA generation how-to. To simplify inferring the sequences to run, the PDB structure preprocessing saves .fasta files with sequence information alongside every preprocessed structure. The script at scripts/data_preprocessing/collect_preprocessed_fastas.py and the consolidate_preprocessed_fastas utility may provide a helpful reference for collating these.
Note
OpenFold3 infers the full sequence of polymers from the pdbx_seq_one_letter_code_can field and _entity_poly_seq records in order to add any unresolved atoms or residues explicitly to the preprocessed structures. This means that any gaps in the structure caused by unresolved residues should have an appropriate spacing in their residue IDs, and the residue ID numbers should match to the residue IDs of the full sequence of the construct incrementally numbered from 1. This works out of the box for PDB structures, but may need special consideration when fine-tuning on custom structures:
Full sequence: M G S S H H H S G L V P R G S H M A S M V E L
Residue IDs: - - - - - - - 8 9 10 11 12 13 14 15 16 17 18 - - 21 22 -
The output will be a single directory with subdirectories containing the alignments for each chain:
pdb_msas_completed/
├── 102l_A
│ ├── bfd_uniref_hits.a3m
│ ├── hmm_output.sto
│ ├── mgnify_hits.sto
│ ├── uniprot_hits.sto
│ └── uniref90_hits.sto
├── 106m_A
│ ├── ...
2.2 MSA Representatives File¶
Since MSAs only need to be generated per unique sequence, we use a “representatives file” to map each MSA directory name to its query sequence. This is used downstream to associate training chains with their corresponding MSA.
Script: scripts/utils/generate_representatives_from_msa_directory.py
Example output MSA_representatives.fasta:
>100d_A
MNIFEMLRIDEGLRLKIYKDTEGYYTIGIGHLLTKSP
>100d_B
SNISRQAYADMFGPTVGDKVRLADTELWIEVEDDLTTAVI
>...
2.3 MSA Preprocessing¶
We convert the raw MSA files into storage- and I/O-efficient npz format to speed up the DataLoader during training.
Script: scripts/data_preprocessing/preparse_alignments_of3.py
Output: per-chain NPZ files* containing the full MSA information:
alignment_cache/
├── 100d_A.npz
├── 100d_B.npz
├── 102l_A.npz
├── ...
3. Dataset Cache Creation¶
3.1 Training Cache¶
The metadata cache generated in PDB preprocessing creates an index of the whole PDB and is subsetted to a final training cache. Following AF3, we apply lightweight filtering (e.g., resolution <= 9.0 A, release date cutoffs), cluster chains and interfaces for balanced sampling during training, and map each chain to its corresponding MSA representative.
Script: scripts/data_preprocessing/create_pdb-weighted_training_dataset_cache.py
Clustering follows AF3 SI §2.5.3:
Protein chains: 40% sequence identity (MMSeqs2)
Peptide chains (< 10 residues): 100% sequence identity
Nucleic acid chains: 100% sequence identity
Small molecule chains: 100% canonical SMILES identity
Interfaces: sorted tuple of individual chain cluster IDs
Output: training_cache.json*
3.2 Validation Cache¶
Similarly, we create a validation set with additional homology filtering against the training set, following AF3 SI §5.8. This applies stricter filtering (resolution <= 4.5 A, token limits) and labels each chain/interface with homology and metric eligibility information. Requires the training cache as input.
Script: scripts/data_preprocessing/create_pdb_validation_dataset_cache.py
Output: validation_cache.json*
4. Templates¶
4.1 Template Structure Preprocessing¶
We preparse the raw potential template structures (typically the full PDB) into npz files for efficient DataLoader access.
Script: scripts/data_preprocessing/preprocess_template_structures_of3.py
Output: individual NPZ files* for each chain in the template pool:
template_structure_arrays/
├── 101m
│ └── 101m_A.npz
│ └── 101m_B.npz
│ └── 101m_C.npz
├── 102l
│ └── 102l_A.npz
├── ...
4.2 Template Cache¶
As the final step, template alignments are preprocessed per dataset (training and validation separately). This creates NPZ files storing template ranks, release dates, and residue-token correspondences, and adds the list of template IDs to the respective dataset cache.
Script: scripts/data_preprocessing/preprocess_template_alignments_new_of3.py
Output: template_cache/* directory and updated training_cache.json* (with template IDs added):
template_cache/
├── 102l_A.npz
├── 103l_A.npz
├── 104l_A.npz
├── ...
The resulting training_cache.json is the final input to the training script.