Understanding Precomputed MSA Handling¶
Here, we aim to provide additional explanations for the inner workings of the MSA components of the OF3 inference pipeline. If you need step-by-step instructions on how to generate MSAs using our OF3-style pipeline, refer to our MSA Generation document. If you need a guide on how to interface MSAs with the inference pipeline, go to the Precomputed MSA How-To Guide.
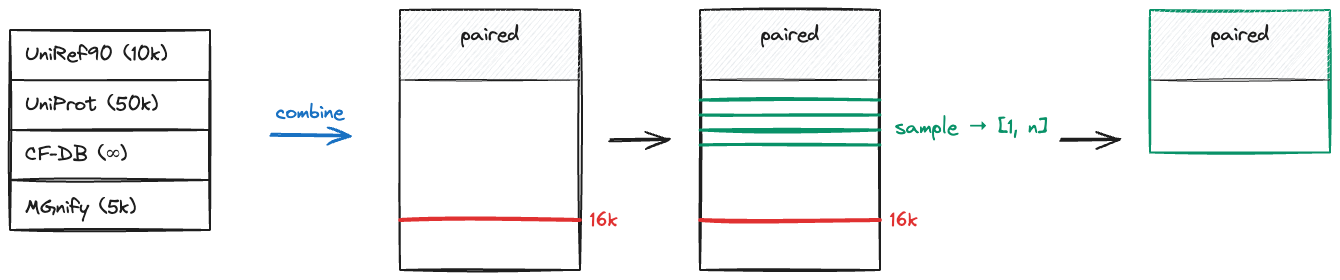
The following diagram provides a high-level overview of the MSA processing pipeline. Per-chain alignments from each sequence database (UniRef90, UniProt, ColabFold DB, MGnify) are combined by vertical concatenation into a single MSA block, capped at 16k sequences. Cross-chain pairing based on species information is then applied to produce the paired MSA rows. The combined MSA is then sampled down to the target size [L_i, nJ], where L_i is the token count (sequence length) of the i-th chain and nJ is the number of sampled MSA rows. During the forward pass, the MSAModuleEmbedder (AF3 Algorithm 8) further subsamples 1024 random rows from this MSA per recycle (controlled by max_subsampled_all_msa).
Specifically, we detail:
1. MSA Input Feature Components¶
Based on the AF3 and AF2-Multimer Supplementary Materials, MSA input features for a single chain are composed of up to 3 components:
query sequence: the protein or RNA sequence whose structure is to be predicted
paired rows: derived from designated MSAs by putting sequences originating from identical species in the same rows
unpaired rows: derived from MSAs by vertically concatenating aligned sequences from all desired sequence database searches; we term the vertical stack of such MSAs the main MSA of the corresponding chain
For multimeric queries, the MSA features for all chains are concatenated horizontally.

Components of OF3 Input MSA features. (left) 5TDH - G protein heterotrimer with GDP, light blue segments indicate gapped parts in the paired MSA, black segments indicate masked parts; (middle) 1OGH - dCTP deaminase homotrimer; 1X1R - M-Ras in complex with GDP and Zn
As shown in the figure above, paired MSAs are only provided for protein chains that are part of complexes with at least two unique protein chains. Besides the query sequences, protein chains in monomeric and homomeric assemblies and RNA chains only get main MSA features, which are treated as implicitly paired for homomers. MSA feature columns for DNA and ligand tokens are empty and masked to prevent their contributions to model activations.
2. MSASettings Reference¶
Users can alter the way MSAs are processed in the OF3 inference pipeline by modifying the MSASettings class via the runner.yml as outlined in the Precomputed MSA How-To Guide.
The 3 main settings to update when using custom precomputed MSAs are:
max_seq_counts: A dictionary specifying how many sequences to read from each MSA file with the associated name. MSA files whose names are not provided in this dictionary will not be parsed. For example, if one wants
uniparc_hits.a3mMSA files to be parsed, the following field should be specified:
dataset_config_kwargs:
msa:
max_seq_counts:
uniprot_hits: 50000
mgnify_hits: 5000
custom_database_hits: 10000
where up to the first 10000 sequences will be read from each uniparc_hits.a3m file.
msas_to_pair: The list of MSA filenames that contain species information that can be used for online pairing. See the Online MSA Pairing section for details.
aln_order: The order in which to vertically concatenate MSA files for each chain for main MSA features. MSA files whose names are not provided in this list will not be used. For example, if one has MSA files named
mgnify_hits,uniprot_hitsanduniparc_hitsand want to vertically concatenate them for each chain in this order, they should update therunner.ymlas follows:
dataset_config_kwargs:
aln_order:
- uniprot_hits
- mgnify_hits
- custom_database_hits
For details on the rest of the settings, see the MSASettings class docstring.
3. Online MSA Pairing¶
Pairing rows of MSAs for heteromeric complexes based on species information is expected to improve the quality of predicted protein-protein interfaces (see this and this publication). When running training or inference on a diverse set of protein complexes like the PDB, protein chains in different complex contexts require different paired MSAs. To avoid having to precompute paired MSAs for a large number of chain combinations, we developed a fast online pairing algorithm, which pairs sequences across MSAs of different chains in the same complex by placing sequences originating from the same species in the same row - the OF3 inference pipeline also accepts precomputed paired MSAs if you want to pair MSAs using your own custom algorithm). See the AF2-Multimer and AF3 Supplementary information for a description of the algorithm or this section in our source code for the exact implementation.
By default, our MSA pipeline uses the UniProt MSAs to generate paired MSAs and so, species information is parsed from UniProt sequence headers. An example sto format is:
# STOCKHOLM 1.0
#=GS sp|P53859|CSL4_YEAST/1-292 DE [subseq from] Exosome complex component CSL4 OS=Saccharomyces cerevisiae (strain ATCC 204508 / S288c) OX=559292 GN=CSL4 PE=1 SV=1
#=GS tr|A6ZRL0|A6ZRL0_YEAS7/1-292 DE [subseq from] Conserved protein OS=Saccharomyces cerevisiae (strain YJM789) OX=307796 GN=CSL4 PE=4 SV=1
#=GS tr|C7GPC7|C7GPC7_YEAS2/1-292 DE [subseq from] Csl4p OS=Saccharomyces cerevisiae (strain JAY291) OX=574961 GN=CSL4 PE=4 SV=1
... rest of the metadata field ...
5k36_I GDPHMACNFQFPEIAYPGKLICPQY--G---------T--E-NK-D-G-------E-D--IIFNYVPGPGTKL----IQ---Y----E--------H---N--G---RT-------------LEAITATL-VGTV-RC---E--E----E----K--KT-DQ-E--E---E---R--EGT-D----Q-S-T--E--E-E-
sp|P53859|CSL4_YEAST/1-292 ----MACNFQFPEIAYPGKLICPQY--G---------T--E-NK-D-G-------E-D--IIFNYVPGPGTKL----IQ---Y----E--------H---N--G---RT-------------LEAITATL-VGTV-RC---E--E----E----K--KT-DQ-E--E---E---R--EGT-D----Q-S-T--E--E-E-
tr|A6ZRL0|A6ZRL0_YEAS7/1-292 ----MACNFQFPEIAYPGKLICPQY--G---------T--E-NK-D-G-------E-D--IIFNYVPGPGTKL----IQ---Y----E--------H---N--G---RT-------------LEAITATL-VGTV-RC---E--E----E----K--KT-DQ-E--E---E---R--EGT-D----Q-S-T--E--E-E-
tr|C7GPC7|C7GPC7_YEAS2/1-292 ----MACNFQFPEIAYPGKLICPQY--G---------T--E-NK-D-G-------E-D--IIFNYVPGPGTKL----IQ---Y----E--------H---N--G---RT-------------LEAITATL-VGTV-RC---E--E----E----K--KT-DQ-E--E---E---R--EGT-D----Q-S-T--E--E-E-
... rest of the alignments ...
and an example a3m format is:
>5k36_I
GDPHMACNFQFPEIAYPGKLICPQY--G---------T--E-NK-D-G-------E-D--IIFNYVPGPGTKL----IQ---Y----E--------H---N--G---RT-------------LEAITATL-VGTV-RC---E--E----E----K--KT-DQ-E--E---E---R--EGT-D----Q-S-T--E--E-E-
>sp|P53859|CSL4_YEAST/1-292
----MACNFQFPEIAYPGKLICPQY--G---------T--E-NK-D-G-------E-D--IIFNYVPGPGTKL----IQ---Y----E--------H---N--G---RT-------------LEAITATL-VGTV-RC---E--E----E----K--KT-DQ-E--E---E---R--EGT-D----Q-S-T--E--E-E-
>tr|A6ZRL0|A6ZRL0_YEAS7/1-292
----MACNFQFPEIAYPGKLICPQY--G---------T--E-NK-D-G-------E-D--IIFNYVPGPGTKL----IQ---Y----E--------H---N--G---RT-------------LEAITATL-VGTV-RC---E--E----E----K--KT-DQ-E--E---E---R--EGT-D----Q-S-T--E--E-E-
>tr|C7GPC7|C7GPC7_YEAS2/1-292
----MACNFQFPEIAYPGKLICPQY--G---------T--E-NK-D-G-------E-D--IIFNYVPGPGTKL----IQ---Y----E--------H---N--G---RT-------------LEAITATL-VGTV-RC---E--E----E----K--KT-DQ-E--E---E---R--EGT-D----Q-S-T--E--E-E-
... rest of the alignments ...
where the first sequence is the query sequence and headers sp|P53859|CSL4_YEAST/1-292, tr|A6ZRL0|A6ZRL0_YEAS7/1-292 and tr|C7GPC7|C7GPC7_YEAS2/1-292 are parsed to get species IDs YEAST, YEAS7 and YEAS2 for the three aligned sequences.
The OF3 pairing code prioritizes sequences that can be paired with as many chains in the complex as possible, over only pairwise-pairable sequences.
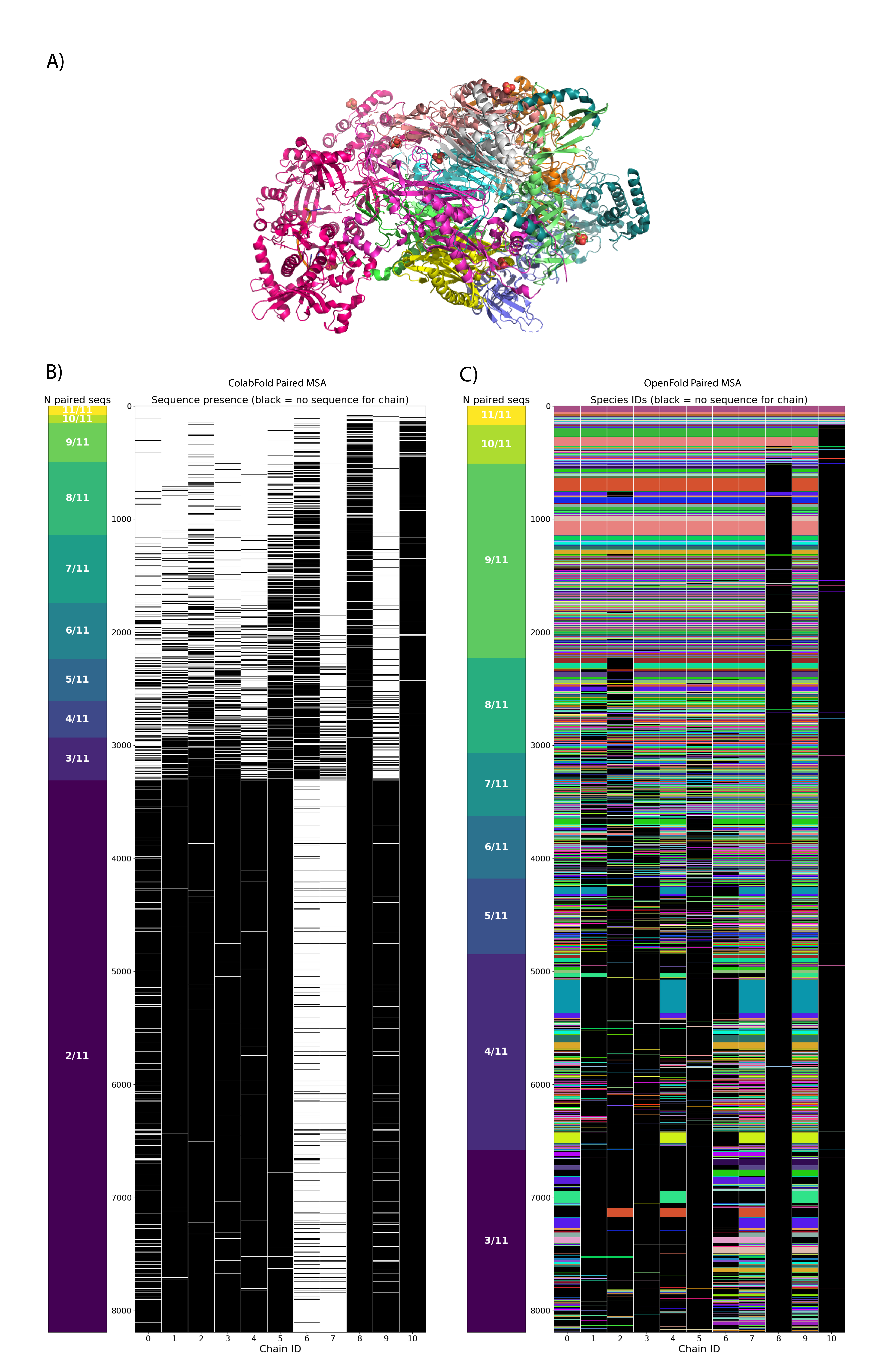
PDB entry 5k36 (A) and comparison of its (B) Colabfold and (C) OpenFold3 paired MSAs. 5k36 is a nuclear exosome complex with 11 protein and 2 RNA chains. The main, wide panels in B) and C) show a simplified representation of the paired MSA, where each row corresponds to a row in the paired MSA, each column corresponds to a chain and each tile indicates which chains in the associated row receive paired sequences (white in CF as no species information is available, colored by species in OF3). Black tiles indicate gapped segments meaning the associated chain does not have a sequence assigned from the species that has sequences for other chains in the same row. The narrow panels indicate how many chains have paired sequences in the corresponding row in the main panels.
4. MSA Reusing Utility¶
Large-scale prediction jobs using OF3 are often done on highly redundant datasets. For example, you may be interested in co-folding a target protein of interest with a library of candidate small molecule drugs, or an antigen of interest with thousands of different antibodies. In these scenarios, the sequence of the target protein does not change across samples and hence, the main MSA for the corresponding chain also remains the same.
In order to reduce the disk space necessary when running predictions, we support reusing the same MSA files for identical chains across different samples. During inference, you can just specify the path to the same MSA files for identical chains in different queries (see the How-To Guide for details on how to do this). For example, given a protein target and 3 different small molecule binders to screen:
Same MSA paths example ...
{
"queries": {
"G-protein-A_GTP": {
"chains": [
{
"molecule_type": "protein",
"chain_ids": "A",
"sequence": "GCTLSAEDKAAVERSKMIDRNLREDGEKAAREVKLLLLGAGESGKSTIVKQMKIIHEAGYSEEECKQYKAVVYSNTIQSIIAIIRAMGRLKIDFGDAARADDARQLFVLAGAAEEGFMTAELAGVIKRLWKDSGVQACFNRSREYQLNDSAAYYLNDLDRIAQPNYIPTQQDVLRTRVKTTGIVETHFTFKDLHFKMFDVGAQRSERKKWIHCFEGVTAIIFCVALSDYDLVLAEDEEMNRMHESMKLFDSICNNKWFTDTSIILFLNKKDLFEEKIKKSPLTICYPEYAGSNTYEEAAAYIQCQFEDLNKRKDTKEIYTHFTCATDTKNVQFVFDAVTDVIIKNNLKDCGLF",
"main_msa_file_paths": "alignments/G-protein-A"
},
{
"molecule_type": "ligand",
"chain_ids": "B",
"ccd_codes": "GTP"
}
]
},
"G-protein-A_GDP": {
"chains": [
{
"molecule_type": "protein",
"chain_ids": "A",
"sequence": "GCTLSAEDKAAVERSKMIDRNLREDGEKAAREVKLLLLGAGESGKSTIVKQMKIIHEAGYSEEECKQYKAVVYSNTIQSIIAIIRAMGRLKIDFGDAARADDARQLFVLAGAAEEGFMTAELAGVIKRLWKDSGVQACFNRSREYQLNDSAAYYLNDLDRIAQPNYIPTQQDVLRTRVKTTGIVETHFTFKDLHFKMFDVGAQRSERKKWIHCFEGVTAIIFCVALSDYDLVLAEDEEMNRMHESMKLFDSICNNKWFTDTSIILFLNKKDLFEEKIKKSPLTICYPEYAGSNTYEEAAAYIQCQFEDLNKRKDTKEIYTHFTCATDTKNVQFVFDAVTDVIIKNNLKDCGLF",
"main_msa_file_paths": "alignments/G-protein-A"
},
{
"molecule_type": "ligand",
"chain_ids": "B",
"ccd_codes": "GDP"
}
]
},
"G-protein-A_GMP": {
"chains": [
{
"molecule_type": "protein",
"chain_ids": "A",
"sequence": "GCTLSAEDKAAVERSKMIDRNLREDGEKAAREVKLLLLGAGESGKSTIVKQMKIIHEAGYSEEECKQYKAVVYSNTIQSIIAIIRAMGRLKIDFGDAARADDARQLFVLAGAAEEGFMTAELAGVIKRLWKDSGVQACFNRSREYQLNDSAAYYLNDLDRIAQPNYIPTQQDVLRTRVKTTGIVETHFTFKDLHFKMFDVGAQRSERKKWIHCFEGVTAIIFCVALSDYDLVLAEDEEMNRMHESMKLFDSICNNKWFTDTSIILFLNKKDLFEEKIKKSPLTICYPEYAGSNTYEEAAAYIQCQFEDLNKRKDTKEIYTHFTCATDTKNVQFVFDAVTDVIIKNNLKDCGLF",
"main_msa_file_paths": "alignments/G-protein-A"
},
{
"molecule_type": "ligand",
"chain_ids": "B",
"ccd_codes": "GMP"
}
]
},
}
}
5. Preparsing Raw MSAs into NPZ Format¶
Two of the main challenges we faced with MSAs were
slow parsing of MSA
stoora3mfiles, which includes the deletion matrix calculationlarge storage costs associated with MSA files
Preparsing raw MSA files into npz files addresses these issues by
moving the per-example numpy array conversion step into an offline preprocessing step that happens only once for each unique MSA
saving the MSA arrays in a compressed format
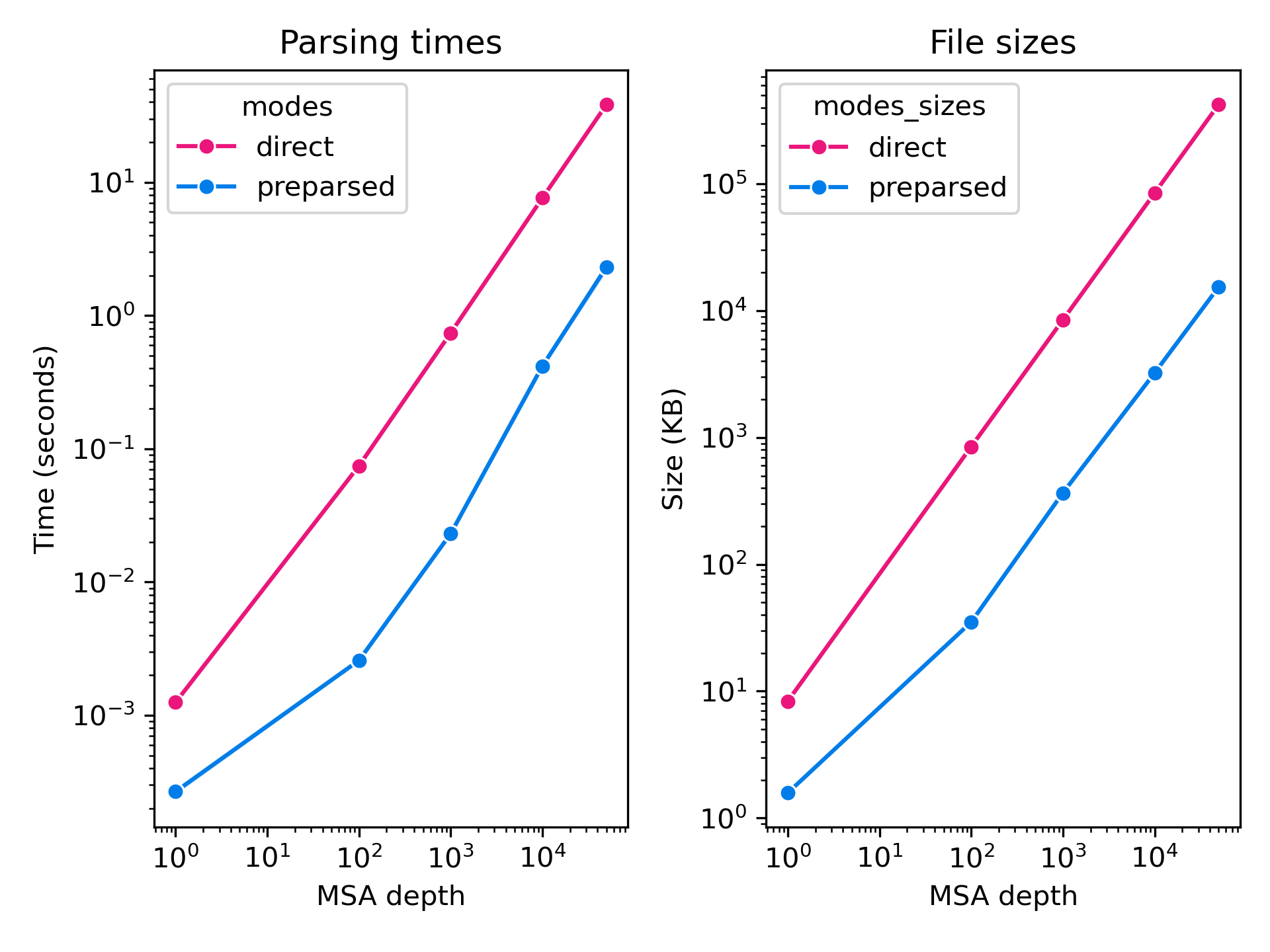
Comparison of sto and npz files: (left) parsing runtimes; (right) file sizes; as a function of number of sequences in the alignments of a protein sequence with 1003 residues.
We found this step to be necessary during training to avoid the online data processing pipeline to bottleneck the model forward/backward passes and to reduce the storage costs associated with our distillation set MSAs.
For inference, preparsing MSAs into npz files can be useful when running large batch jobs on highly redundant datasets, for example when screening one or a few target protein against a library of small molecule ligands or antibodies.
MSAs can be preparsed using the preparse_alignments_of3.py script given that they are provided in the format outlined in the Precomputed MSA How-To Guide. The resulting npz files will contain a dictionary mapping file names to pre-parsed MsaArray objects, which store the MSAs in a numpy array format, alongside its deletion matrix and metadata required for online pairing and can be used directly by the inference data pipeline.
{
"uniprot_hits": MsaArray(...),
"mgnify_hits": MsaArray(...),
"custom_database_hits": MsaArray(...),
}