Understanding Dataset Caches¶
The data preprocessing pipeline produces several JSON cache files that serve as the interface between raw PDB data and the training script. This page explains their structure and contents.
Metadata Cache¶
The PDB preprocessing script produces a metadata.json that indexes all preprocessed structures. The idea of this index is to do the preprocessing once, then be flexible with subsetting to train or validation set splits after. It contains two top-level dictionaries: structure_data and reference_molecule_data.
Structure Data¶
Each entry in structure_data describes a single preprocessed structure at the entry, chain, and interface level. The core datapoint unit of AlphaFold3 is an individual chain or interface, which is what is sampled during training. The resulting structure shown to the model is typically a crop centered on a random atom of that chain/interface.
The keys in the chains dictionary (e.g. "1", "2") are the numerical chain IDs assigned during structure preprocessing, which replace the original PDB label_asym_id/auth_asym_id. These numerical IDs are also the chain identifiers used in the preprocessed AtomArrays.
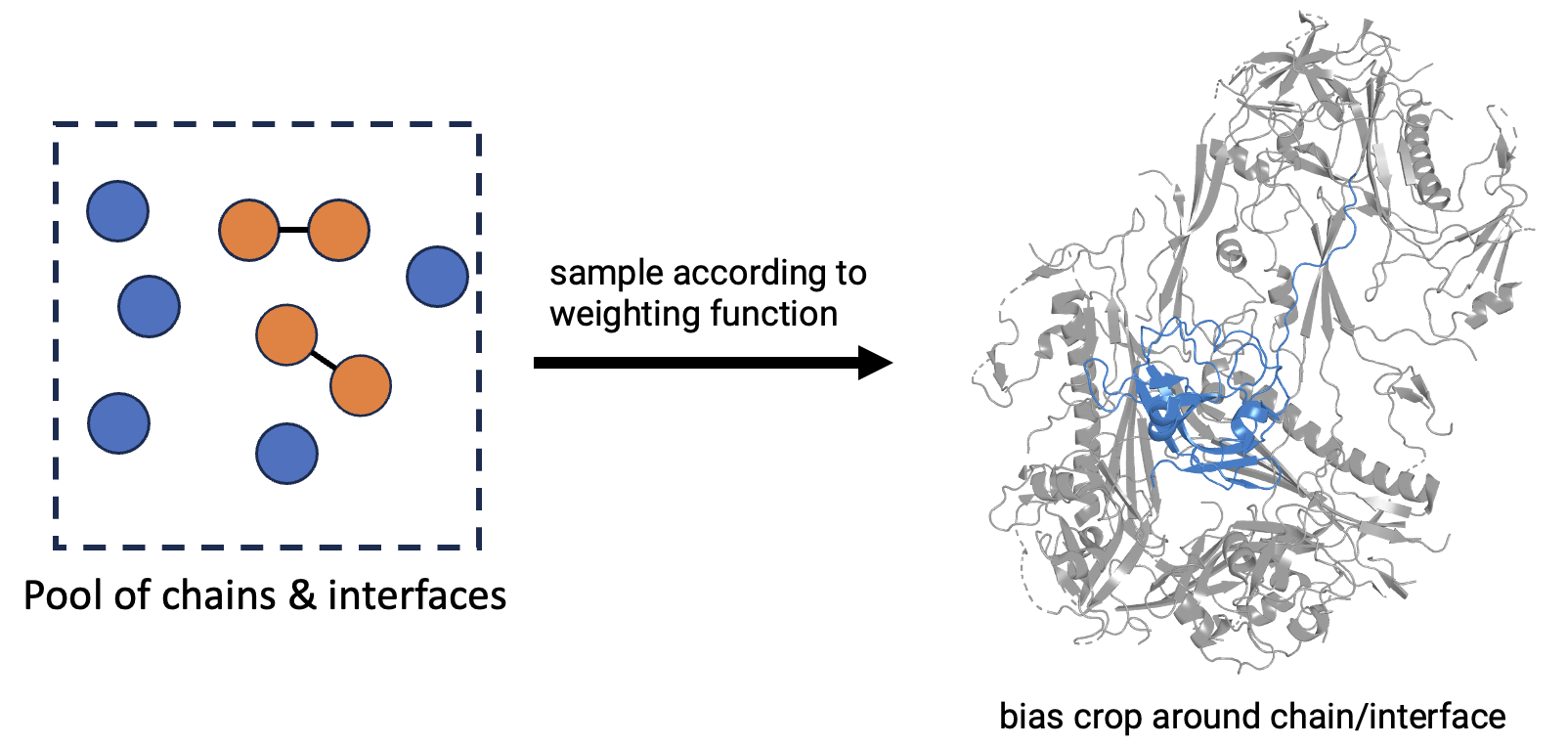
The formal specification is defined in format.py. Example entry:
"4ws7": {
"release_date": "2015-07-15",
"status": "success",
"resolution": 1.88,
"experimental_method": "X-RAY DIFFRACTION",
"token_count": 612,
"chains": {
"1": {
"label_asym_id": "A",
"auth_asym_id": "A",
"entity_id": 1,
"molecule_type": "PROTEIN"
},
"2": {
"label_asym_id": "B",
"auth_asym_id": "A",
"entity_id": 2,
"molecule_type": "LIGAND",
"reference_mol_id": "5UC"
}
},
"interfaces": [
["1", "2"]
]
}
Field |
Description |
|---|---|
Key (e.g. |
Unique structure identifier. Must match the folder/file names in |
|
Release date, used for time-based train/val splitting. |
|
Processing outcome: |
|
Resolution in Angstrom (NaN for NMR). Used for filtering. |
|
E.g. |
|
Total tokens in the structure. Used to cap validation set size. |
|
Matches the |
|
One of |
|
Ligand-only. Points to an entry in |
|
Pairs of chain IDs with minimum heavy-atom separation < 5 Angstrom. |
Reference Molecule Data¶
Each unique ligand encountered during preprocessing gets an entry in reference_molecule_data, alongside a corresponding SDF file in reference_molecules/.
AlphaFold3 generates random 3D conformers for every residue and ligand during training using RDKit. These conformers inform the model about molecular geometry, chirality, and hybridization (the model has no explicit stereochemistry embedding). The preprocessing step tries multiple conformer generation strategies and caches the one which succeeded, so the DataLoader can skip directly to it during training. (Note that we still regenerate conformers dynamically during training!)
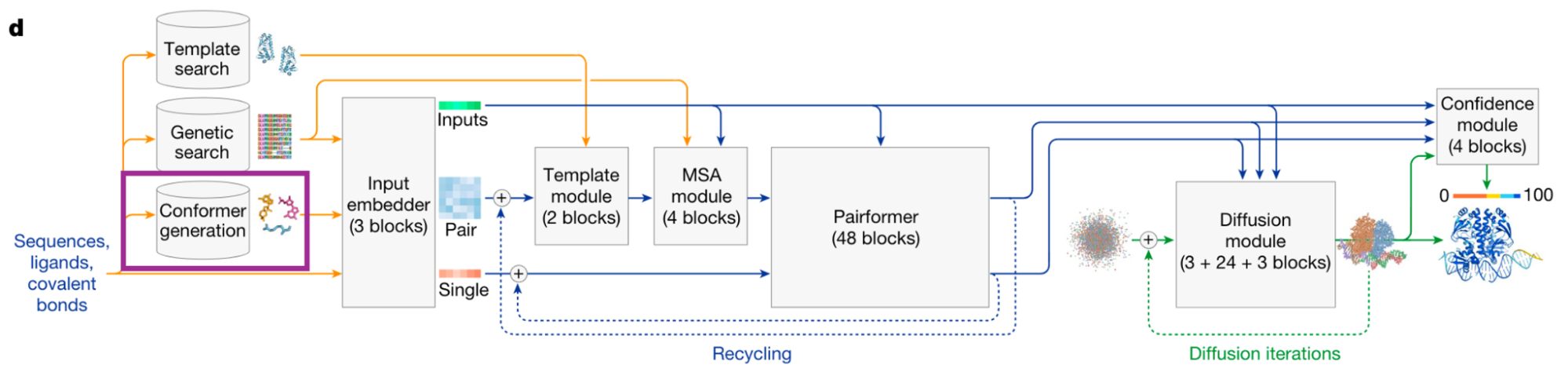 AlphaFold3 architecture from Abramson et al.; the conformer generation step is highlighted in purple.
AlphaFold3 architecture from Abramson et al.; the conformer generation step is highlighted in purple.
The four strategies, tried in order:
"default": Standard RDKit ETKDG conformer generation."random_init": Random coordinate initialization instead of ETKDG (helps for larger molecules)."use_fallback": Idealized CCD coordinates (pdbx_model_Cartn_*_ideal)."use_fallback"(variation): Model-derived CCD coordinates if idealized ones are missing.
Example entries (a standard monomeric ligand and a disaccharide glycan):
"ATP": {
"residue_count": 1,
"conformer_gen_strategy": "default",
"fallback_conformer_pdb_id": null,
"canonical_smiles": "Nc1ncnc2c1ncn2[C@@H]1O[C@H](CO[P@@](=O)(O)O[P@](=O)(O)OP(=O)(O)O)[C@@H](O)[C@H]1O"
},
"2pvw_2": {
"residue_count": 2,
"conformer_gen_strategy": "default",
"fallback_conformer_pdb_id": null,
"canonical_smiles": "CC(=O)N[C@H]1[C@H](O[C@H]2[C@H](O)[C@@H](NC(C)=O)CO[C@@H]2CO)O[C@H](CO)[C@@H](O)[C@@H]1O"
}
Field |
Description |
|---|---|
Key |
CCD code for monomeric ligands, or |
|
Number of residues (1 for standard ligands, >1 for glycans). |
|
Which strategy succeeded (see above). |
|
PDB-ID associated with model-derived fallback coordinates. |
|
RDKit-canonicalized SMILES. Used for ligand clustering. |
Note
Glycans (multi-residue ligands): The handling of multi-residue ligands such as glycans is ambiguous in the AlphaFold3 SI. In OpenFold3, we link them together into single ligand molecules instead of separating the individual monomeric residues, to be consistent with the treatment of other covalent ligands. Unlike monomeric ligands which are keyed by their CCD code, glycans are keyed by [PDB-ID]_[entity-ID] (e.g. "2pvw_2") and have residue_count >1. This technically means glycan reference molecules are not unique across structures, which causes some negligible overhead in preprocessing but does not matter at training time.
SDF File Format¶
Each reference molecule is saved as an SDF file in the reference_molecules/ output folder. These use standard V2000 molfile format with two custom annotation fields appended. Example for acetate (ACT.sdf):
RDKit 3D
4 3 0 0 0 0 0 0 0 0999 V2000
0.8140 -0.0385 0.0035 C 0 0 0 0 0 0 0 0 0 0 0 0
1.5232 -1.0718 0.0134 O 0 0 0 0 0 0 0 0 0 0 0 0
1.5226 1.1766 -0.0223 O 0 0 0 0 0 1 0 0 0 0 0 0
-0.6545 -0.0200 0.0167 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 2 0
1 3 1 0
1 4 1 0
M CHG 1 3 -1
M END
> <atom_annot_atom_name> (1)
C O OXT CH3
> <atom_annot_used_atom_mask> (1)
True True True True
$$$$
The custom annotations are:
atom_annot_atom_name: Canonical atom names for this ligand, matching the names in the preprocessed AtomArrays.atom_annot_used_atom_mask: A per-atom mask.Truefor all atoms when a conformer was successfully generated. When CCD-deposited fallback coordinates were used, any atoms with missing coordinates are markedFalse.
Training Cache¶
The training cache (training_cache.json) selects and enriches a subset of the metadata cache for training. It adds clustering information (for balanced sampling) and MSA/template pointers.
Script: scripts/data_preprocessing/create_pdb-weighted_training_dataset_cache.py
The example below shows the final form of a training cache entry, after template IDs have been added by the template preprocessing step:
"1a3n": {
"release_date": "1998-04-29",
"resolution": 1.8,
"chains": {
"1": {
"label_asym_id": "A",
"auth_asym_id": "A",
"entity_id": 1,
"molecule_type": "PROTEIN",
"reference_mol_id": null,
"alignment_representative_id": "7pch_A",
"template_ids": [
"2lhb_A",
"1vhb_A",
"1flp_A",
"1ash_A",
"1eca_A"
],
"cluster_id": "12193",
"cluster_size": 821
},
"2": {
"label_asym_id": "E",
"auth_asym_id": "A",
"entity_id": 3,
"molecule_type": "LIGAND",
"reference_mol_id": "HEM",
"alignment_representative_id": null,
"template_ids": null,
"cluster_id": "42685",
"cluster_size": 9925
}
},
"interfaces": {
"1_2": {
"cluster_id": "12193_42685",
"cluster_size": 791
}
}
}
Additional fields compared to the metadata cache:
Field |
Description |
|---|---|
|
Name of the corresponding MSA NPZ file in the alignment cache. |
|
Template chain IDs to sample from during training. Added by the template preprocessing step; |
|
Cluster assignment for this chain/interface. |
|
Number of members in the cluster. |
Training Cache Reference Molecule Data¶
The reference_molecule_data section in the training cache carries over all fields from the metadata cache and adds one:
"ATP": {
"conformer_gen_strategy": "default",
"fallback_conformer_pdb_id": null,
"canonical_smiles": "Nc1ncnc2c1ncn2[C@@H]1O[C@H](CO[P@@](=O)(O)O[P@](=O)(O)OP(=O)(O)O)[C@@H](O)[C@H]1O",
"set_fallback_to_nan": false
}
Field |
Description |
|---|---|
|
Set to |
Validation Cache¶
The validation cache (validation_cache.json) follows the same structure as the training cache but with additional homology and quality labels for proper evaluation. It is created by comparing against the training set.
Script: scripts/data_preprocessing/create_pdb_validation_dataset_cache.py
Key differences from the training cache:
Requires the training cache as input for homology comparison
Stricter filtering: resolution <= 4.5 Angstrom, token limits
Homology detection: 40% sequence identity threshold, 0.85 Tanimoto similarity for ligands
Two subsets: multimer and monomer validation sets
Example entry (subset of 7vl5 showing one protein chain, one ligand chain, and their interface):
"7vl5": {
"release_date": "2022-03-09",
"resolution": 1.93,
"token_count": 1463,
"chains": {
"1": {
"label_asym_id": "A",
"auth_asym_id": "A",
"entity_id": 1,
"molecule_type": "PROTEIN",
"reference_mol_id": null,
"alignment_representative_id": "7vl6_A",
"template_ids": [
"5gsl_A",
"5gsl_B",
"6jow_A"
],
"cluster_id": "2024",
"cluster_size": null,
"low_homology": true,
"metric_eligible": true,
"use_metrics": false,
"ranking_model_fit": null,
"source_subset": "base",
},
"3": {
"label_asym_id": "C",
"auth_asym_id": "A",
"entity_id": 2,
"molecule_type": "LIGAND",
"reference_mol_id": "BOG",
"alignment_representative_id": null,
"template_ids": null,
"cluster_id": "5086",
"cluster_size": null,
"low_homology": false,
"metric_eligible": false,
"use_metrics": false,
"ranking_model_fit": 0.9312,
"source_subset": "base",
}
},
"interfaces": {
"1_3": {
"cluster_id": "2024_5086",
"cluster_size": null,
"low_homology": true,
"metric_eligible": true,
"use_metrics": false,
"source_subset": "base"
}
}
}
Additional fields compared to the training cache:
Field |
Description |
|---|---|
|
Whether this chain/interface has low sequence homology to the training set (below 40% identity for proteins, below 0.85 Tanimoto for ligands). |
|
Whether this chain/interface is eligible for metric computation. Requires low homology and, for ligand chains, sufficient model ranking fit. |
|
Whether this chain/interface is actually used for reporting metrics. A subset of |
|
Ligand model quality score from RCSB (0-1). |
|
Which validation subset this entry belongs to: |